METHYLMALONIC ACIDEMIA (MMA)
DİĞER İSİMLERİ:
- Isolated MMA
- Methylmalonic aciduria
GENEL BİLGİ:
Metilmalonik asidemi vücutta belirli protein ve lipitlerin parçalanması için gereken enzimlerin bazı mutasyonlar sonucu düzgün bir şekilde çalışamamasıyla ortaya çıkar. Bu durum bazı amino asitlerin ve yağ asitlerinin doğru bir şekilde çalışmasını engeller. Bu hastalık kalıtsaldır. Genellikle yeni doğanlarda veya erken çocukluk döneminde görülür. Etkileri hafif şiddetli seyredebilirken ölümcül de olabilir.
BELİRTİLER / BULGULAR:
Çocuklarda kusma, sıvı kaybı, zayıf kas tonusu (hypotonia), gelişim geriliği, aşırı yorgunluk, vücutta asit-baz dengesizliği, karaciğer büyümesi (hepatomegaly), kilo kaybı ve bazı hastalarda yüksek seviyelerde amonyak birikimi ya da felç görülür. Uzun vadede beslenme problemleri, zekâ geriliği, kronik böbrek rahatsızlığı, pankreas iltihaplanması (pancreatitis) ile karşılaşılabilir. Eğer tedavi edilmezse ilerleyen aşamalarda koma ve hatta ölümle sonuçlanabilir.
GÖRÜLME SIKLIĞI:
Metilmalonik asidemi hastalığının yaklaşık olarak her 50.000 yeni doğan bebekten birinde görüldüğü kaydedilmiştir. Fakat Türkiye’deki görülme sıklığı henüz tam olarak bilinmemektedir.
GENETİK DEĞİŞİKLİKLER / ETKEN FAKTÖRLER:
MMUT, MMAA, MMAB, MMADHC, ve MCEE genlerindeki mutasyonların bu hastalığa neden olduğu bilinmektedir. Etkileri mutasyona uğrayan gen ile mutasyonun şiddetine bağlıdır. MMA sahibi bireyler iki gruba ayrılabilir:
- Sadece metilmalonik asidin artış gösterdiği izole olmuş MMA
- Metilmalonik asidin yanı sıra homosistein artışı bulunan MMA
Birinci gruptaki bozukluğa MMAA, MMAB ve MMUT genlerindeki mutasyonlar neden olur. Bu gruptaki vakaların neredeyse yarısı MMUT genindeki mutasyonlar sonucu oluşmuştur. Bu gen birçok aminoasidin (proteinlerin yapıtaşı), lipitlerin ve kolesterolün parçalanmasından sorumlu metilmalonil-CoA mutaz enziminin üretilmesini sağlar. Bu enzim B12 vitamini (cobalamin) ile beraber çalışır. MMUT genindeki mutasyonlar bu enzimin yapısını değiştirir ya da miktarını azaltır. Bu nedenle aminoasitler, lipitler ve kolesterolün düzgün bir şekilde parçalanması engellenir. Sonuç olarak metilmalonil-CoA maddesi ve diğer bazı toksik bileşimler vücutta ve organlarda birikerek bu hastalığın semptomlarının ve belirtilerinin oluşmasına neden olur.
MMUT genindeki mutasyonlar sonucu oluşan metilmalonik asidemi hastalığı genel olarak iki çeşittir. Bu gendeki mutasyonlar sonucu enzimler aktifliğini kaybediyorsa buna MUT0 denir ve MUT0 metilmalonik asideminin en ciddi ve en az sonuç alınan tipidir. Eğer bu mutasyonlar enzim aktivitesini bozmuyorsa bu MUT– diye adlandırılır. Bu iki grup arasındaki ayrım kesin değildir ama uzmanların çoğu hücrelere aşırı miktarda B12 eklendiğinde enzim aktivitesinin artması durumunda hastalığın MUT– grubuna ait olduğunda hemfikir. Ayrıca MUT– klinik olarak daha hafif bir forma sahiptir.
MMAA, MMAB ya da MMADHC genlerindeki mutasyonlar da metilmalonik asideminin oluşmasına neden olur. Cobalamin A (cblA) tipi metilmalonik asidemi MMAA genindeki mutasyonlarla ortaya çıkar. Cobalamin B tipi metilmalonik asidemiye MMAB genindeki mutasyonlar neden olur. Bu iki durum da MUT0 tipi ile benzerdir. Bununla birlikte, cblA ve cblB hastalarının çoğu bir çeşit B12 vitamini (hydroxycobalamin) desteği aldığında klinik ve metabolik olarak ilerleme gösteriyor.
MMAA ve MMAB enzimlerinin vücuttaki asıl görevleri tam olarak bilinmiyor. Bu enzimler metilmalonil-CoA mutaz enziminin düzgün bir şekilde çalışması için gerekli olan başka bir çeşit B12 vitamininin (adenosycobalamin) üretimine yardım ediyor olabilirler.
İkinci gruptaki bozukluğa sahip hastalarda metilmalonik asit ve homosistein artışı gözlenir. Bunlar Cobalamin C (cblC), Cobalamin D (cblD) ve Cobalamin F (cblF) eksikliğini de kapsar. En yaygın olanı cblC eksikliğidir. Bu tipte MMADHC geni değişime uğrar. Bu durumdaki hastalarda vücut sıvısındaki metilmalonik asit ve homosistein miktarı artar. Bu durum da bazı görme bozuklukları ve nörolojik problemlerin oluşmasına neden olabilir. CblD ve cblF ile onların sorumlu olduğu genler olan MMADHC ve LMBRD1 kaynaklı MMA çok nadir görülen durumlardır.
Metilmalonik asidemiye neden olan bir başka durum da MCEE genindeki mutasyonlardır. Bu gen de metilmalonil-CoA epimeraz enzimi için gerekli talimatları verir. Metilmalonil-CoA mutaz enzimi gibi bu enzim de aminoasit, lipit ve kolesterolün yıkımında rol alır. Farkları metilmalonil-CoA epimeraz enzimi metilmalonik asideminin daha hafif bir tipinin oluşmasına neden olur.
Bunlardan farklı olarak, henüz tespit edilmemiş başka genler de metilmalonik asidemi hastalığına neden olabilir.
Hastalığın kalıtımı otozomal çekinik genlerle sağlanır. Yani hastalığın ortaya çıkması için ilgili genin (MUT, MMAA, MMAB, MMADHC, MCEE, LMBRD1) her iki kopyasında da mutasyon ya da değişimlerin olması gerekir. Genelde, değişmiş genin bir kopyası anneden bir diğeri babadan gelir. Dolayısıyla hasta çocuğun ebeveynleri taşıyıcıdır. Taşıyıcı bireyler hastalığın semptomlarını ve belirtilerini göstermezler. Vücut kromozomlarıyla taşındığı için kız ve erkek çocuklarda oluşma ihtimali eşittir.

GÖRSEL: Otozomal çekinik karakterin kalıtımı
KAYNAK: U.S. NATIONAL LIBRARY of MEDICINE
Taşıyıcı olan ebeveynler; %25 ihtimalle hastalık genini taşımayan çocuklara, %50 ihtimalle taşıyıcı çocuklara ve %25 ihtimalle hasta çocuklara sahip olabilir.
Genetik ya da nadir hastalıklarda teşhis genelde zordur. Sağlık çalışanları teşhis koyabilmek için öncelikle hastanın sağlık geçmişine, semptomlarına, fiziksel muayenesine ve laboratuvar test sonuçlarına bakar.
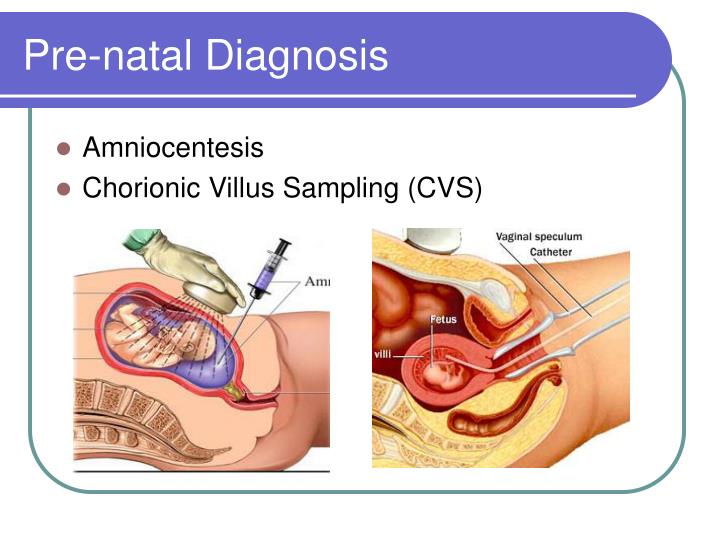
Hastanın ve ailenin sağlık geçmişinin detaylı incelenmesi, ayrıntılı klinik değerlendirmeler ve çeşitli özel testler sonucu yaşamın ilk haftalarında hastalık teşhis edilebilir. MMA genellikle hamilelikte amniyotik sıvıdaki metilmalonik asit miktarı ölçülerek, sıvıdaki enzim aktivitesine bakılarak veya fetüs / uterustan
alınan doku örnekleriyle (amniyosentez / koryon villus biyopsisi -CVS-) teşhis edilebilir. Amniyosentezde, fetüsün çevresini saran amniyotik sıvıdan doku örnekleri alınır ve analiz edilir. CVS de ise fetüs 10-12 haftalık olduğunda plasentadan alınan bir doku örneği incelenir.
- Amniyosentez
- Koryon Villus Biyopsisi

Kaynak: https://image3.slideserve.com/6595796/pre-natal-diagnosis-n.jpg
{kind=link}
Maalesef hastalığı tamamen yok etmek için henüz bir tedavi yok. Yalnızca etkilerini azaltmak ve ilerlemesini yavaşlatmak için bazı tedavi yöntemleri uygulanabilir.
Metilmalonik asidemi hastalığının tedavisinde düşük protein yüksek kalorili diyetler uygulanır. Bu diyetler çocuklarda dikkatli takip edilmelidir. İzolösin, valin, treonin ve metiyonin gibi aminoasitler içeren beslenme düzeninden kaçınılmalıdır. Çünkü bu aminoasitler vücutta metilmalonik aside dönüşür.
İlaç tedavisinde enjeksiyonla cobalamin (b12 vitamini), antibiyotik ve karnitin (bir tür aminoasid) takviyesi yapılır. Hastalık ilerledikçe tedavi organ nakline kadar gidebilir. İlaç ve beslenme tedavileri her hasta için özeldir.
KAYNAKLAR:
- https://rarediseases.org/rare-diseases/acidemia-methylmalonic/
- https://ghr.nlm.nih.gov/condition/methylmalonic-acidemia#resources
- https://www.genome.gov/19016900/
- https://emedicine.medscape.com/article/1161799-overview
- https://rarediseases.info.nih.gov/diseases/7033/methylmalonic-acidemia
- https://www.kelseygroup.com/mma-disease-brief-overview/
- http://www.behcetuzdergisi.com