Furkan M. TORUN & Dr. Oktay I. KAPLAN
Teşhisi ve tedavisi en zor hastalıklar hangileridir dendiği zaman tıbbi eğitim alanlar dahi çoğu kimsenin aklına Joubert sendromu, Bardet-Biedl sendromu gibi nadir hastalıklar gelmez. Geçtiğimiz 100 yılda ve özellikle son 30 yılda tıbbi alanlarda kayda değer gelişmeler yaşanmasına rağmen nadir hastalıkların teşhisi ve tedavisinde hala zorlanmaktayız.
Nadir hastalıkları kısaca tanımlamak gerekirse Avrupa Birliğinin aldığı karara göre her 2.000 kişide bir, ABD’deki karara göre ise 200.000 ‘den az sayıda kişiyi etkileyen hastalıklara ‘nadir hastalık’ ismi verilir. – Dünyada şu an tanımlanan en az 7000 farklı nadir hastalık olduğu bilinmekte iken bu 7000 civarı hastalığın %95’i için hâlâ Amerikan Gıda ve İlaç Birliği (FDA) tarafından onaylanmış bir tedavi yöntemi bulunmamaktadır. Nadir hastalıkların adından da anlaşılacağı üzere bu hastalıklar tek başlarına çok küçük bir popülasyonu etkilese de Dünya’da ve Türkiye’de sırasıyla 350 milyon ve 5 milyon kişinin bu hastalıklardan etkilendiği tahmin edilmektedir.
Göz rengi, saç biçimi, boyumuzun uzunluğu gibi bireysel özelliklerimiz için kılavuzluk eden ve canlılığın en temel ihtiyaçlarından biri olan proteinlerin kodlanmasında görev alan DNA’mızdaki küçük parçalara gen adı verilir. Gen terapisi/tedavisi ise temelinde, fonksiyonel bozukluğa ya da eksikliğe sahip olan bir genin sebep olduğu hastalığı tedavi etmek amacıyla hastaya, söz konusu gene ait doğru DNA parçasının verilmesi işlemidir. Kısacası gen terapisi, sorunları nükleotid düzeyinde DNA ameliyatı ile çözmektedir. Nadir hastalıkların çoğunun (%80) gen bozukluğundan kaynaklandığını düşünürsek hastalıkların tedavisi için dışarıdan eksik olan genin verilmesi akıllı bir çözüm gibi görünmektedir.
Uzun zamandır üzerinde çalışılan gen terapi yöntemi bu noktada sorunlara çözüm olma potansiyeline sahip miydi? 7000’e yakın farklı hastalığın her biri için farklı bir sürece gereksinim duyulması nadir hastalıklar için çalışma yapmayı zorlaştırıyor olsa da gen terapisinin arka planındaki en büyük avantajlardan birisi de kalıcı çözüm sunma potansiyeline sahip olmasıdır.
Toz pembe hayal gibi görünen bu rüyanın aslında günümüzde uygulanma noktasına ulaştığına dikkat çektikten sonra bugünkü yazımıza, gen terapisinin tarihçesi ve uygulamalı örnekleri hakkında konuşarak devam edeceğiz. Teorik olarak yöntem başarılı gözüksede gen terapisinin geçmişine bakıldığında ilk denemelerin yapıldığı dönemlere göre epey bir mesafe kat edildiği fakat bu süreçte bazı kayıpların da yaşandığı görülmektedir.
1960’lı yıllarda bakterilerde moleküler genetiğin ve gen transferinin temellerinin oluşturulması, daha sonraki yıllarda viral vektörlerle ya da genetik olarak modifiye edilmiş hücreler sayesinde diğer canlılara gen transferi yapılması kaçınılmaz hale getirilmiştir. 1972 yılında ise Science dergisinde yayınlanan makale ile insan genetik hastalıklarında gen terapisi çözümüne bir kez daha değinilmiştir. Fakat gen terapisinin/tedavisinin temelinde yer alan sorun, ilgili genin hücrenin içine nasıl verileceğinin bilinmemesiydi. Takvimler 1980’leri gösterdiğinde retrovirüs vektörlerinin gelişmesi, memeli hücrelerine yapılacak olan gen transferinin de önünü açmıştı.
Bu bağlamda ilk girişim Martine Cline tarafından 1980 yılında gerçekleştirilmeye çalışılmış olsa da daha sonra iki β-talasemi hastasının kemik iliği tedavisi almasından ötürü M. Cline’a yapılan destek, insanlar üzerinde rekombinat DNA araştırması ve deneylerini düzenleyen kuralları ihlal ettiği gerekçesiyle NIH (National Institues of Health [Ulusal Sağlık Enstitüsü]) tarafından kesilmiştir.
İlerleyen süreçte gen terapisi fikrinin tedavi amaçlı insanlarda ilk kullanımı ise 14 Eylül 1990’da French Anderson ve takım arkadaşlarının yaptığı çalışmaya dayanmaktadır. Bu çalışma sayesinde ADA (adenosine deaminase) genini taşıyan retrovirüs vektörü ile şiddetli/ağır kombine immün yetmezlik (AKİY [ing.: SCID]) isimli hastalığı taşıyan Ashanthi DeSilva ve Cindy Kisik isimli 2 çocuğun tedavisi sağlanmıştır. Bu yapılan çalışmada yürütülen aşamalar ise kısaca beyaz kan hücrelerinin hasta çocuktan alınması, Adenosine deaminase’yi üreten fonksiyonel genin bu alınan hücrelere yerleştirilmesi ve doğrulanan hücrelerin tekrar hastaya verilmesidir. Ne yazık ki bu başarıyı, gen tedavisinin doğrudan bir sonucu olarak ölen 18 yaşındaki Jesse Gelsinger izledi. Üzücü olay, OTC (ornithine transcarbamylase) genini doğrudan kan dolaşımına yerleştirmek için adenovirüsün in vivo olarak hastaya verildiği bir gen terapisinde gerçekleşmiştir. Jesse Gelsinger, tedaviden sonraki bir kaç gün içerisinde muhtemelen vücudun virüse karşı şiddetli olarak verdiği immün sistem cevabı sonucu oluşan çoklu organ yetmezliğinden öldü. Aslında bu durumun en büyük sebeplerinden birisi de genlerin teslimi için kullanılan vektörlerin genomun rastgele bir yerine girmesidir ve bu bağlamda gen terapisi hakkında birçok soru işareti bireylerin zihninde oluşmaya devam etmiştir. Bu durum gen terapisinde zaman içerisinde bir çok inişlerin ve çıkışların olmasına sebep olmuştur.

Cindy ve Ashanthi 1992’de gen terapisinin öncüleri ile beraber: (soldan sağa doğru) French Anderson, MD; Michael Blaese, MD; ve Kenneth Culver.
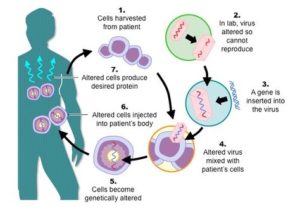
1. Hücrelerin hastadan seçilmesi
2. Laboratuvar koşullarında virüs için çeşitli değişikliklerin yapılması
3. İlgili genin virüse enjekte edilmesi
4. Değiştirilen virüsün hastanın hücrelerine gönderilmesi
5. Hücrelerin genetik olarak modifiye olması
6. Değişime uğrayan hücrelerin hastaya verilmesi
Gen terapisinin mucizevi dokunuşuna bir başka örnek ise X kromozomunda yer alan ILG2RG geninin mutasyona uğraması sonucu ortaya çıkan SCID’nin bir başka formu olan SCID-X1 (X-linked severe combined immunodeficiency) hastalığı, bir başka ismiyle, Bubble Baby Syndrome olarak isimlendirilen Balon Bebek Sendromu verilebilir. Bu hastalıkta, bağışıklık sisteminde olan sorun nedeniyle belki biz sağlıklı bireylerde bir gribe neden olabilecek çevresel faktörler bu hastalığa sahip bireylerde ölümcül etkiye sahip olabilir. Bu yüzden SCID hastalığına sahip bireylerde yaşam süresi maalesef çok kısadır. İlerleyen süreçte, gen terapisi ile IL2RG geninin doğru formu hastalara vektör aracılığıyla verilmiştir. Ne yazık ki, yapılan bir çalışmada tedavi edilmiş olan 9 hastanın 4’ünde tedaviden 31-68 ay sonra lösemi gözlenmiştir.

Yazımızın başında genlerin hasta bireyin hücrelerine teslimatının en önemli problemlerden biri olduğunu ve bunu sağlamak için (gene delivery) virüslerin kullanıldığından bahsetmiştik. Doğru formu verilecek olan genin iletilmesinde en önemli faktörlerden biri olan ideal vektörün seçimi yapılırken; bağışıklık sistemi tarafından cevap oluşturmama (toksik olmama), teslim edilecek bölgeye/dokuya spesifik olma, verilecek DNA için yeterli kapasiteye sahip olma gibi bazı özellikler aranmaktadır.
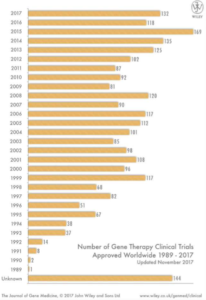
Gen terapisine dair istatistiklere değinecek olursak, ülkeler bazındaki karşılaştırmasına bakıldığında en çok klinik gen terapisinin Amerika Birleşik Devletlerinde yapıldığı görülmektedir. Ek olarak, klinik tedavi boyutunda en çok gen terapisi görmüş hastalık türü %65’lik bir oran ile kanserdir. Kanseri takip eden diğer hastalık türü ise %11 ile tek genden kaynaklanan (monogenik) hastalıklardır.15 İstatistiksel olarak büyük tabloya dikkat etmek gerekirse, gen terapisinin yıllar içerisinde onaylanmış klinik denemelerinin sayısı epey dalgalanma yaşamış olsa da sonuç itibariyle gün geçtikçe bu sayı artmıştır.
Gen terapisinin emekleme yıllarında meydana gelen üzücü olaylar sonraki yıllarda o eksiklikleri giderme yoluyla aşılmaya çalışıldı ve son dönemlerde gen terapisi adına büyük gelişmeler yaşandı. Gen terapisi için üretilen ve onaylanan ilk ilaç, uniQure isimli şirketin ürünü olan ve 1.1 milyon € (euro) gibi çok uçuk bir fiyata sahip olan Glybera idi. Milyonda bir kişiyi etkileyen ve LPL genindeki mutasyondan kaynaklanan LPLD (Lipoprotein Lipase Deficiency [Lipoprotein Lipaz Eksikliği]) hastalığını hedef alan Glybera ilacının 25 Ekim 2017’de bitmiş olan onay tarihi de pazar payının büyük olmaması sebebiyle yenilenmedi.
Bir başka gen terapi çalışması da en önemli duyu organlarımızdan biri olan gözlerimiz için gerçekleştirildi. Spark Therapeutics isimli şirketin ürettiği FDA (U.S. Food and Drug Administration [ABD Gıda ve İlaç İdaresi]) onaylı ilaç olan Luxturna (voretigene neparvovec-rzyl), RPE65 geninde mutasyona sahip olan bireylerin görsel pigmentlerinde bozulmaya ve fotoreseptörlerin işlev kaybına neden olarak bireylerde görme duyusunu etkileyen bir hastalığı hedef aldı. Çeşitli sıçan ve köpekler üzerinde yapılan çalışmalardan elde edilen sonuçlar heyecan yaratmış ve AAV virüsü ile göz anatomisinin insana benzerlik göstermesinden ötürü Briard türü köpeklerde yapılan denemelerde olumlu sonuçlar alınmıştır ve dikkat çekici bir şekilde, tüm denemelerde hastalar, tedaviden birkaç ay sonra çeşitli derecelerde olsa da, görsel düzelmelerin birkaç yönünü sergilemiştir.
Gen terapisine dair bir başka örnek olarak da 2012 yılında farelerde yapılan ve başarıyla sonuçlanan bir başka çalışma verilebilir: Vücudumuzun hemen hemen bütün hücrelerinde bulunan görme, koku alma gibi bir çok hayati fonksiyonda rol alan silya isimli yapılar bulunmaktadır ve bu çalışmada silya isimli yapılarda meydana gelen mutasyonlar veya çeşitli sorunlar sonucu ortaya çıkan siliyopati isimli hastalık grubu ele alınmıştır. IFT88 isimli, silyalarda görev alan bir gendeki mutasyon sonucu farelerde koku alma duyusunda hasar meydana gelmiştir ve bu durum farelerin erkenden ölmelerine sebep olmaktadır. Bu durumu çözmek isteyen araştırmacılar, bir virüs çeşidi olan adenovirüs içerisine fonksiyonel olarak görevini gerçekleştirebilen IFT88 genini yerleştirip daha sonra bu virüsü farelere enjekte etmişlerdir. Genin görevini yerine getirmesiyle, farelerde koku fonksiyonun kazanımı sağlanmıştır.
Yakın zamanda ülkemizde de gündem olan ve gen terapisi ile artık çözüme kavuşabilen bir diğer hastalık ise SMA (Spinal Muskuler Atrofi). Omurilikte bulunan ve kasların hareketinden sorumlu olan bazı hücrelerin görevlerini yerine getirmemesi veya zarar görmesi sonucu ortaya çıkan bu hastalık, kol, bacak ve gövde kaslarının hareket edememesi ile sonuçlanır. 4 farklı tipi olan bu hastalık, kaslarda zayıflık gözlenirken, bağımsız oturamama ve baş kontrolünü dahi yapamayacak duruma gelme ile de karşılaşılabilir. –
New England Journal of Medicine adlı özel bir dergide yayınlanan çalışma ile SMA Tip 1 hastalığına neden olan SMN1 genine adeno-associated virus serotype 9 (AAV9) isimli bir virüs aracılığı ile fonksiyonel olan gen 15 hastaya tek seferde teslim edildi. Üç hasta düşük dozda alırken, 12 hastaya ise yüksek doz uygulandı. Çalışmanın ilk fazında, yüksek doz alan hastaların motor fonksiyonlarında iyileşme gözlendi ve eski hallerine göre daha az ihtiyaç duyacak duruma geldiler. Hatta çalışmanın sonunda yüksek doz alan hastaların çok büyük kısmı baş kontrolünü sağlayabilirken, 2 hasta ise emeklemeyi başarmıştı.
Yazımızın en başında gen terapisinin nadir hastalıklar için birer umut ışığı olduğundan kısaca bahsetmiştik. Zira şu ana kadar gerçekleştirilmiş yayınlar ve yapılan klinik çalışmaların verilerinin tutulduğu ClinicalTrials veritabanındaki kayıtların sayısı da bu konuda bizlere birer dayanak noktası olmuş durumda.
2017 gen terapisi açısından belki de başarılı yıl idi
Araştırmacılar, 2017 Mart ayında Fransa’da gerçekleşen gen terapisi çalışması ile kan hücrelerini etkileyen orak hücre hastalığının (sickle-cell disease) pençesinde olan bir genci iyileştirmeyi başardıklarını duyurdu. Çocuğun kemik iliğindeki kök hücrelerine ulaşmayı başaran bilim insanları, kırmızı kan hücrelerinin orak şeklini almasını engellemek için ilgili genin kopyasını alınan hücrelere girmesini sağlayarak hücrelerde genetik değişiklik yaptılar. Tedavi edilen hücreler hastaya geri verildiğinde normal kan hücreleri yapmaya başlamıştır. Tedavinin ardından geçen 2 yıldan fazla bir sürede hasta, bu sağlık probleminin yan etkilerinden kurtulmak için yeterince sağlıklı alyuvara (kırmızı kan hücresine) sahip olmuştur
Bir başka benzer çalışma ise epidermolizis bülloza (epidermolysis bullosa) isimli hastalığa sahip ve cildinin dış katmanının %60’ına yakınını kaybeden bir çocuk için gerçekleştirildi. Araştırmacılar bu duruma çözüm bulmak adına çocuğun cildinden sağlıklı olan hücreleri aldılar, bu duruma sebep olan LAMB3 geninin fonksiyonel olan versiyonu ile laboratuvar şartlarında alınan hücreleri genetik olarak modifiye ettiler ve bu hücrelerin küçük katmanlara dönüşmesine izin verdiler. Daha sonrasında bu katmanlar üç ameliyatlık bir dizi operasyonla Almanya’daki bir hastanede çocuğa nakledildi.
Gen terapisi alanında yapılan son araştırmalar ve deneyler ise F8 (faktor 8) genindeki değişikliklerden kaynaklanan kan pıhtılaşması için gerekli proteini üretemeyen ve kontrol edilemeyen kanamalara sebep olan Hemofili A hastaları için de bir umut ışığı oldu. BioMarin isimli biyoteknoloji şirketi tarafından desteklenen ve aralık ayında The New England Journal of Medicine dergisinde yayınlanan makale ile 9 erkek hasta üzerinde bir buçuk yıl süren çalışma ile hastaların daha az kanama sorunu yaşadığı ve pıhtılaşma faktörlerinde de pozitif yönde ilerlemeler sağlandığı kanıtlanmıştır. Benzer semptom ve belirtiler gösteren ama F9 genindeki değişikliklerden kaynaklanan Hemofili B hastaları için de gen terapisi yine 2017 yılında yapıldı. 10 erkek Hemofili B hastası üzerinde yürütülen bu çalışmada birçok klinik denemede de olduğu gibi yine adeno-associated viral (AAV) vektörü kullanıldı ve bu sefer karaciğer için özel hazırlanmış bir gen bölgesini(promoteri) ve F9 transgenini (bir organizmadan başka bir organizmaya sokulan DNA segmenti) iletmek için kullanıldı.
Disiplinlerarası bir yaklaşım olan gen terapisi; teknoloji, biyoloji ve tıp alanındaki gelişmelerden etkilenerek gün geçtikçe daha verimli ve daha az yan etki gösterir hale gelecek iken nadir hastalıklar için ise hikayenin yeni başladığı ve daha çok yolun alınacağı söylenebilir.
KAYNAKLAR:
“Rare diseases – European Commission.” https://ec.europa.eu/health/rare_diseases/overview_en. Erişim tarihi: 28 Nis. 2018.
“Office of Special Medical Programs > Office of Orphan Products … – FDA.” 30 Kas. 2017, https://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProductsandTobacco/OfficeofScienceandHealthCoordination/ucm2018190.htm. Erişim tarihi: 28 Nis. 2018.
“NRD-1008 FactSheet_5 – National Organization for Rare Disorders.” http://cdn.rarediseases.org/wordpresscontent/wp-content/uploads/2014/11/NRD-1008-FactSheet_5.pdf. Erişim tarihi: 28 Nis. 2018.
“Gene Therapy for Human Genetic Disease? | Science.” http://science.sciencemag.org/content/178/4061/648. Erişim tarihi: 23 Nis. 2018.
“An early history of gene transfer and therapy. – NCBI.” https://www.ncbi.nlm.nih.gov/pubmed/8049304. Erişim tarihi: 23 Nis. 2018.
“Martin Cline loses appeal on NIH grant | Science.” http://science.sciencemag.org/content/218/4567/37.1. Erişim tarihi: 23 Nis. 2018.
“Gene therapy finds its niche | Nature Biotechnology.” 7 Şub. 2011, https://www.nature.com/articles/nbt.1769. Erişim tarihi: 23 Nis. 2018.
“T Lymphocyte-Directed Gene Therapy for ADA− SCID: Initial … – Science.” http://science.sciencemag.org/content/270/5235/475. Erişim tarihi: 23 Nis. 2018.
“Fatal systemic inflammatory response syndrome in a … – Science Direct.” https://www.sciencedirect.com/science/article/pii/S1096719203001690. Erişim tarihi: 1 May. 2018.
“[Gene therapy of SCID-X1]. – NCBI.” https://www.ncbi.nlm.nih.gov/pubmed/18046520. Erişim tarihi: 30 Nis. 2018.
“Why Gene Therapy Caused Leukemia In Some ‘Boy In … – ScienceDaily.” 10 Ağu. 2008, https://www.sciencedaily.com/releases/2008/08/080807175438.htm. Erişim tarihi: 30 Nis. 2018.
“Insertional oncogenesis in 4 patients after retrovirus-mediated gene ….” 7 Ağu. 2008, https://www.jci.org/articles/view/35700. Erişim tarihi: 30 Nis. 2018.
“Gene therapy clinical trials worldwide to 2017 – Wiley Online Library.” 25 Mar. 2018, https://onlinelibrary.wiley.com/doi/abs/10.1002/jgm.3015. Accessed 30 Jul. 2018.
“First Gene Therapy Drug Approved in Europe Set to Launch, Priced at ….” https://globalgenes.org/raredaily/first-gene-therapy-drug-approved-europe-set-launch-priced-u-s-1-4-million/. Erişim tarihi: 1 May. 2018.
“Familial lipoprotein lipase deficiency – Genetics Home Reference – NIH.” https://ghr.nlm.nih.gov/condition/familial-lipoprotein-lipase-deficiency. Erişim tarihi: 1 May. 2018.
“Advances in Gene Therapy for Diseases of the Eye | Human Gene ….” https://www.liebertpub.com/doi/full/10.1089/hum.2016.040. Erişim tarihi: 1 May. 2018.
“Gene therapy restores sense of smell to mice : Nature News & Comment.” 4 Eyl. 2012, https://www.nature.com/news/gene-therapy-restores-sense-of-smell-to-mice-1.11333. Erişim tarihi: 5 May. 2018.
“Gene therapy rescues cilia defects and restores olfactory … – Nature.” 2 Eyl. 2012, https://www.nature.com/articles/nm.2860. Erişim tarihi: 5 May. 2018.
“Spinal Muscular Atrophy | Muscular Dystrophy Association.” https://www.mda.org/disease/spinal-muscular-atrophy. Accessed 30 Jul. 2018.
“Spinal muscular atrophy – Genetics Home Reference – NIH.” https://ghr.nlm.nih.gov/condition/spinal-muscular-atrophy. Accessed 30 Jul. 2018.
“Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy ….” https://www.nejm.org/doi/full/10.1056/NEJMoa1706198. Accessed 30 Jul. 2018.
“ClinicalTrials.gov.” https://clinicaltrials.gov/. Accessed 30 Jul. 2018.
“Gene Therapy in a Patient with Sickle Cell Disease | NEJM.” 2 Mar. 2017, http://www.nejm.org/doi/full/10.1056/NEJMoa1609677. Erişim tarihi: 5 May. 2018.
“Regeneration of the entire human epidermis using transgenic … – Nature.” 8 Kas. 2017, https://www.nature.com/articles/nature24487. Erişim tarihi: 5 May. 2018.
“Hemophilia – Genetics Home Reference – NIH.” 24 Jul. 2018, https://ghr.nlm.nih.gov/condition/hemophilia. Accessed 30 Jul. 2018.
“AAV5–Factor VIII Gene Transfer in Severe Hemophilia A | NEJM.” 9 Ara. 2017, https://www.nejm.org/doi/10.1056/NEJMoa1708483. Erişim tarihi: 5 May. 2018.
“Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX ….” https://www.nejm.org/doi/full/10.1056/NEJMoa1708538. Accessed 30 Jul. 2018.İçerik Görsel Kaynağı: https://pixabay.com/tr/dna-biyoloji-bilim-dna-sarmal-163710/